新型功能材料能够实现从清洁能源到信息处理等技术应用的根本性突破1,2,3,4,5,6,7,8,9,10,11。____从微芯片到电池和光伏发电,无机晶体的发
新型功能材料能够实现从清洁能源到信息处理等技术应用的根本性突破1,2,3,4,5,6,7,8,9,10,11。____从微芯片到电池和光伏发电,无机晶体的发现一直受到昂贵的试错方法的瓶颈。与此同时,随着数据和计算的增加,语言、视觉和生物学的深度学习模型展示了新兴的预测能力12,13,14。在这里,我们展示了大规模训练的图网络可以达到前所未有的泛化水平,从而将材料发现的效率提高一个数量级。以持续研究中发现的48,000个稳定晶体为基础15,16,17,效率的提高使得能够在当前凸包下方发现220万个结构,其中许多结构逃过了人类之前的化学直觉。我们的工作代表了人类已知的稳定材料的数量级扩展。最终凸包上的稳定发现将可用于筛选技术应用,正如我们对层状材料和固体电解质候选物的演示一样。在稳定结构中,有736个已通过独立实验实现。数以亿计的第一原理计算的规模和多样性也解锁了下游应用的建模能力,特别是导致高度准确和强大的学习原子间势,可用于凝聚相分子动力学模拟和高保真零-离子电导率的射击预测。
主要的
能量有利的无机晶体的发现在固态化学中具有基础科学和技术意义。几十年来的实验方法已经在无机晶体结构数据库(ICSD)15,18中编目了20,000个计算稳定的结构(总共200,000个条目)。然而,由于成本、吞吐量和合成复杂性,这种策略对于规模化来说是不切实际的19。相反,材料项目(MP)16、开放量子材料数据库(OQMD)17、AFLOWLIB20和NOMAD21倡导的计算方法使用基于密度泛函理论(DFT)的第一性原理计算作为物理能量的近似值。根据我们自己的重新计算22、23、24(参见方法),研究人员将从头计算与简单替换相结合,将计算稳定的材料改进为48,000种。尽管人们一直在寻求有助于进一步材料发现的数据驱动方法,但到目前为止,机器学习技术在估计竞争相能量凸包的稳定性(分解能)方面无效25。
在本文中,我们通过大规模主动学习扩大了用于材料探索的机器学习,产生了第一个能够准确预测稳定性的模型,从而可以指导材料发现。我们的方法依赖于两个支柱:首先,我们建立生成不同候选结构的方法,包括新的对称感知部分替换(SAPS)和随机结构搜索26。其次,我们使用最先进的图神经网络(GNN)来改进给定结构或成分的材料属性的建模。在一系列轮次中,这些用于材料探索的图网络(GNoME)接受可用数据的训练,并用于过滤候选结构。过滤后的候选者的能量是使用DFT计算的,既验证模型预测,又充当数据飞轮,以便在下一轮主动学习中在更大的数据集上训练更鲁棒的模型。
通过这个迭代过程,GNoME模型发现了超过220万个相对于之前的工作稳定的结构,特别是包含计算和实验结构的聚合数据集15,16,17,27。鉴于已发现的材料争夺稳定性,更新后的凸包由381,000个新条目组成,总共421,000个稳定晶体,代表了之前所有发现的一个数量级的扩展。与机器学习其他领域的观察结果一致28,我们观察到我们的神经网络预测随着数据量的增加呈幂律提高。最终的GNoME模型准确预测了11meV原子-1的能量,并将结构稳定预测的精度(命中率)提高到80%以上,仅使用成分每100次试验提高33%,而之前的工作为1%17。此外,这些网络发展出新兴的分布外泛化。例如,GNoME能够准确预测具有5个以上独特元素的结构(尽管在训练中遗漏了),提供了有效探索该化学空间的首批策略之一。我们通过将预测与实验和更高保真度的r2SCAN(参考文献 29)计算进行比较来验证研究结果。
最后,我们证明GNoMEdiscovery中生成的数据集为下游应用程序解锁了新的建模功能。结构和松弛轨迹提供了一个庞大且多样化的数据集,使得能够以前所未有的精度和零样本泛化来训练学习的等变原子间势30、31。我们通过分子动力学模拟估计离子电导率来证明这些潜力对于材料性能预测的前景。
生成和过滤概述
可能材料的空间太大,无法以公正的方式进行采样。由于没有可靠的模型来廉价地近似候选能量,研究人员通过化学直觉限制生成来指导搜索,通过替换相似离子或枚举原型来完成22。尽管提高了搜索效率17,27,但这种策略从根本上限制了候选者的多样性。通过神经网络引导搜索,我们能够使用多样化的方法来生成候选者,并在不牺牲效率的情况下对晶体空间进行更广泛的探索。
为了生成和过滤候选者,我们使用两个框架,如图1a所示。首先,通过修改现有晶体来生成候选结构。然而,我们通过调整离子取代概率来大力增强取代集,以优先发现发现,并使用新提出的对称感知部分取代(SAPS)来有效地实现不完全取代32。此次扩展导致超过109名候选人在主动学习过程中;由此产生的结构通过GNoME进行过滤,使用基于体积的测试时间增强和通过深度集成的不确定性量化33。最后,对结构进行聚类并对多晶型物进行排序,以使用DFT进行评估(参见方法)。在第二个框架中,成分模型在没有结构信息的情况下预测稳定性。输入是简化的化学式。通过氧化态平衡生成通常过于严格(例如,忽略Li15Si4)。使用宽松的约束(参见方法),我们使用GNoME过滤组合,并初始化100个随机结构,以便通过从头随机结构搜索(AIRSS)26进行评估。在这两个框架中,模型提供能量预测,并根据竞争相的相对稳定性(分解能量)选择阈值。评估是通过维也纳从头算仿真包(VASP)34中的DFT计算进行的,我们与材料项目16相比,测量了发现的稳定材料的数量以及预测的稳定材料的精度(命中率)。
图1:GNoME实现高效发现。
图1
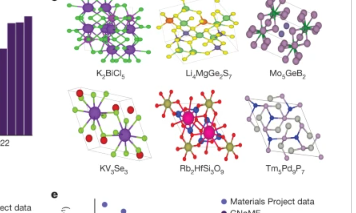
a,基于GNoME的发现的总结展示了基于模型的过滤和DFT如何充当数据飞轮来改进预测。b,GNoME实现的探索已经产生了381,000种新的稳定材料,几乎比之前的工作大了一个数量级。c、736个结构已被独立实验验证,其中示出了6个示例50、51、52、53、54、55。d,图网络预测的改进使得能够有效地发现材料的组合区域,例如,具有六个独特的元素,即使训练集停在四个独特的元素上。e,当对随机结构搜索的域外输入进行测试时,GNoME展示了新兴的泛化能力,表明通用能源模型的进展。
全尺寸图像
侏儒
所有GNoME模型都是预测晶体总能量的GNN。通过元素的单热嵌入将输入转换为图表。我们遵循消息传递公式35、36,其中聚合投影是具有快速非线性的浅多层感知器(MLP)。对于结构模型,我们发现通过整个数据集中原子的平均邻接度来标准化从边缘到节点的消息非常重要。初始模型是根据2018年材料项目的快照进行训练的,其中包含约69,000种材料。之前的工作以28meV原子-1的平均绝对误差(MAE)为基准对这项任务进行了基准测试(参考文献 37);然而,我们发现改进的网络实现了21meV原子-1的MAE。我们修复了这个有前景的架构(参见方法),并在本文的其余部分重点关注扩展。
主动学习
我们加速材料发现框架的核心步骤是主动学习。在结构和组成框架中,使用GNoME过滤的候选结构是使用DFT计算和材料项目中的标准化设置进行评估的。松弛结构产生的能量不仅验证了晶体结构的稳定性,而且还被纳入迭代主动学习工作流程中,作为候选生成的进一步训练数据和结构。尽管结构框架和组合框架的命中率一开始分别低于6%和3%,但通过六轮主动学习,性能稳步提高。GNoME模型的最终集成在松弛结构上的预测误差提高到11meV原子-1,命中率分别大于80%和33%,清楚地显示了规模的好处。图1d提供了最终GNoME命中率的分析。
缩放法则和泛化
GNoME模型的测试损失性能随着进一步的数据呈现出幂律的改进。这些结果符合深度学习中的神经尺度定律28、38,并表明进一步的发现工作可以继续提高泛化能力。需要强调的是,与语言或视觉的情况不同,在材料科学中,我们可以继续生成数据并发现稳定的晶体,这些晶体可以重复使用以继续扩大模型。我们还通过测试基于图1e中随机搜索26产生的晶体替换数据训练的结构模型,展示了对分布外任务的新兴泛化。与我们的结构管道(通过替换,包含接近最小值的结构)生成的数据相比,这些例子通常是高能局部最小值并且不分布。尽管如此,我们观察到规模上的明显改善。这些结果表明,最终的GNoME模型是向社区提供通用能量预测器迈出的重要一步,能够通过深度学习处理不同的材料结构。
发现稳定晶体
使用所描述的扩展深度学习材料探索的过程,我们将已知稳定晶体的数量增加了几乎一个数量级。特别是,GNoME模型发现220万个晶体结构对于材料项目来说是稳定的。其中,381,000个条目作为新发现的材料存在于更新的凸包上。
与其他有关结构预测的文献一致,未来的发现可能会将GNoME材料从凸包上剔除,类似于GNoME如何取代材料项目和OQMD中的至少5,000种“稳定”材料。有关改进已发现组合物结构的讨论,请参阅补充说明1。尽管如此,无花果。图1和图2总结了稳定材料,图1b重点关注随时间的增长。我们在图2a中看到具有四个以上独特元素的结构数量大幅增加。这是特别有希望的,因为这些材料已被证明对于以前的发现工作来说是困难的27。我们的缩放GNoME模型克服了这一障碍,并能够在组合大区域中进行有效发现。
声明本站所有作品图文均由用户自行上传分享,仅供网友学习交流。若您的权利被侵害,请联系我们